Diagnostic notes | Non refereed |
Pre-harvest food safety diagnostics for Salmonella serovars. Part 2: Molecular diagnostics
Wondwossen Abebe Gebreyes, DVM, PhD, Diplomate ACVPM
Swine Health and Production Medicine, North Carolina State University, College of Veterinary Medicine, Department of Farm Animal Health and Resources Management, Raleigh, North Carolina; Tel: 919-513-6141; Fax: 919-513-6383; E-mail: wagebrey@unity.ncsu.edu.
Keywords: diagnosis, food safety, pre-harvest, salmonellaCite as: Gebreyes WA. Pre-harvest food safety diagnostics for Salmonella serovars. Part 2: Molecular diagnostics. J Swine Health Prod 2003;11(3):141-145. Also available as a PDF.
Search the AASV web site for pages with similar keywords.
Several molecular methods have in-creasingly become the preferred tools for diagnosis of foodborne pathogens, including Salmonella. These methods have allowed tracking of foodborne pathogens at the population level by enhanced detection and subtyping. Methods based on genotyping tend to have a high discriminatory power and offer rapid and sensitive subtyping, complementing conventional approaches that are based on phenotype, for example, serotyping and phage typing. Additional factors for consideration in the choice of molecular methods include reproducibility, ease of interpretation, and time and cost efficiency.
Not all molecular approaches are equally suitable for all foodborne pathogens. Selection of an appropriate method depends on the presence or absence of the specific gene allele of interest, stability or hypervariability of genomes in some pathogens, and the objectives of the molecular analysis. For example, genotypic methods, such as pulsed-field gel electrophoresis (PFGE) in association with serotyping, are often used to discriminate Salmonella serovars in foodborne outbreaks and epidemiological studies.
Three major molecular approaches used for the diagnosis and epidemiology of Salmonella serovars will be discussed: amplification of gene allele(s) by polymerase chain reaction (PCR), including multiplex PCR and real-time PCR; DNA-DNA hybridization (Southern blot); and fingerprinting (geno-typing), using methods based on restriction fragments, amplification, or both.
PCR
The basic principle of PCR is amplification (up to a million times) of the specific DNA sequence of interest within a few hours. The products of amplification (amplicons) are separated by size using gel electrophoresis. A detailed demonstration of PCR techniques can be found at http://www. dnalc.org/shockwave/pcranwhole.html.1
Before PCR may be used for diagnostic purposes, it is essential to have prior knowledge of a unique DNA sequence in the target (suspect) organism, which might be specific to a genus, species, or strain. In some cases, even if the same DNA sequence is present in two organisms, PCR may be used to discriminate between them on the basis of DNA fragment size.
Polymerase chain reaction has been used to detect Salmonella serovars in samples from different sources, such as poultry,2 swine,3 and cattle.4 The genes being detected are usually virulence determinants, such as sipB/sipC,5 himA,6 and fimA and tctC genes.7
Pathogenic Salmonella serogroups may be differentiated using PCR to identify unique regions of the rfb gene cluster, which encodes for O-antigen biosynthesis. The gene rfbJ is unique to serogroup B, which includes serovar Typhimurium, and rfbS and rfbE are unique to serogroup D, which includes serovar Typhi.8,9
A PCR method that can efficiently discriminate between Salmonella serovars is currently being developed, in which a DNA-based assay identifies serovar-specific antigens. First, the alleles encoding for three phases of the flagellar (H) antigen, fliC, fljB and flpA, are sequenced and characterized. Unique DNA markers are then identified, and these results are combined with similar assays for O antigens to complete the molecular diagnostic assay.10 Another recent report describes a PCR assay that uses the 16S and 23S ribosomal spacer region to discriminate between Salmonella serovars.11
Multiplex PCR
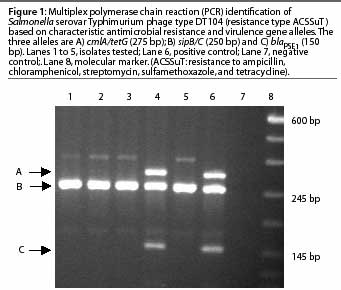
Serovar Typhimurium has recently been identified as the most common Salmonella serovar isolated from humans and swine in the United States.12,13 An important characteristic of this serovar is the emergence of multi-drug resistant phenotypes, which are often characterized by phage typing. Phage type DT104, one of the most common multi-drug resistant and pandemically distributed phage types, is commonly resistance- (R-) type ACSSuT, exhibiting resistance to ampicillin, chloramphenicol, streptomycin, sulfamethoxazole, and tetracycline. Phage type DT193, also of public health importance and often associated with swine products,3,12 is commonly R-type AKSSuT, exhibiting resistance to ampicillin, kanamycin, streptomycin, sulfamethoxazole, and tetracycline.
The resistance genes involved in the pentaresistant DT104 and DT193 phage types have been previously described.3,14 These phage types can be detected using a multiplex PCR, which amplifies multiple genes in a single reaction. As an example, a multiplex PCR has been developed5 that detects phage type DT104 by amplifying two virulence determinant primers and one or more of the antimicrobial resistance genes described (Figure 1).
Real-time PCR
Real-time PCR is similar to conventional PCR in its basic principle and method, but measures accumulation of a fluorescent probe.15 The signal increases in direct proportion to the amount of PCR product in the reaction, enabling detection while the reaction is progressing. Two important advantages of real-time PCR are achievement of shorter processing time and lower cost by eliminating post-amplification gel analysis and enabling quantitative analysis of gene products. The latter is particularly useful in gene expression assays, where it is necessary to amplify cDNA in order to quantify it after it is produced from mRNA in the reverse-transcriptase reaction. Other advantages of real-time PCR are increased throughput, reduced chances of carryover contamination, and elimination of post-PCR processing as a potential source of error. Non-specific chemical agents that intercalate with double-stranded DNA, such as SYBR-green, are commonly used in singleplex reactions of real-time PCR. The real-time PCR principle has also been extended to multiplex reactions using sequence-specific probes, for example, the Taqman probe system16 and hairpin-shaped oligonucleotide probes called molecular beacons.6 As the molecular beacon approach is highly discriminatory and specific, it enables detection of as few as 2 colony forming units per PCR reaction. This method is often used to detect single nucleotide polymorphisms,17 for example, the gyrA gene mutation of fluoroquinolone resistance, that may produce adverse phenotypic manifestations.
DNA-DNA hybridization (Southern blot)
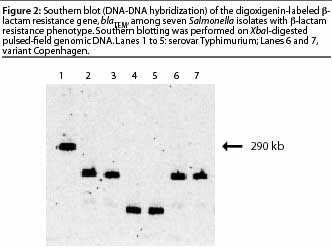 Hybridization of nucleic acid is one of the preferred approaches to confirm
identification of a nucleic acid sequence in an
organism. The blotting method suitable for
identifying DNA sequences in bacterial foodborne pathogens is known as a Southern blot
or DNA-DNA hybridization. Genomic or plasmid DNA from the organism is
digested using one or more restriction enzymes, and digestion fragments are
separated using agarose gel electrophoresis. After single-stranded fragments are
blotted onto nitrocellulose or nylon paper, the gene probe of interest (labeled with
radioactive or chemiluminescent digoxigenin) is hybridized. The target DNA sequence
can be identified and its fragment size measured using an apparatus to detect
radioactivity or chemiluminescence. Several studies have identified specific gene alleles
in Salmonella. In an experiment to detect
the [beta]-lactam resistance gene,
blaTEM, and its location in seven
Salmonella isolates, Southern blotting (DNA-DNA
hybridization) was performed on
XbaI-digested pulsed-field genomic DNA hybridized
using a digoxigenin-labeled
blaTEM gene probe (Figure 2). The blot showed that
the blaTEM gene was located in different
DNA fragments, showing that the isolates carrying the gene were not clonal; and that
the gene was present on a large fragment (290 kb) in one isolate, implying that it was
located on the chromosome.
Hybridization of nucleic acid is one of the preferred approaches to confirm
identification of a nucleic acid sequence in an
organism. The blotting method suitable for
identifying DNA sequences in bacterial foodborne pathogens is known as a Southern blot
or DNA-DNA hybridization. Genomic or plasmid DNA from the organism is
digested using one or more restriction enzymes, and digestion fragments are
separated using agarose gel electrophoresis. After single-stranded fragments are
blotted onto nitrocellulose or nylon paper, the gene probe of interest (labeled with
radioactive or chemiluminescent digoxigenin) is hybridized. The target DNA sequence
can be identified and its fragment size measured using an apparatus to detect
radioactivity or chemiluminescence. Several studies have identified specific gene alleles
in Salmonella. In an experiment to detect
the [beta]-lactam resistance gene,
blaTEM, and its location in seven
Salmonella isolates, Southern blotting (DNA-DNA
hybridization) was performed on
XbaI-digested pulsed-field genomic DNA hybridized
using a digoxigenin-labeled
blaTEM gene probe (Figure 2). The blot showed that
the blaTEM gene was located in different
DNA fragments, showing that the isolates carrying the gene were not clonal; and that
the gene was present on a large fragment (290 kb) in one isolate, implying that it was
located on the chromosome.
DNA fingerprinting (genotyping)
Fingerprinting, also referred to as genotyping, identifies an organism or strain on the basis of its nucleic acid content (most commonly DNA). By definition, the most sensitive and accurate way of fingerprinting is DNA sequencing. However, approaches that indicate variation in sequences are less costly and more rapid than sequencing, and have been used effectively. Genotyping methods are commonly based on identification of restriction fragments, for example, by PFGE; amplification by PCR, for example, by amplified fragment length polymorphism (AFLP) or repetitive sequence PCR (Rep-PCR); or DNA sequence, for example, by multi-locus sequence typing (MLST).
Restriction fragment-based genotyping methods: PFGE
The basic principle of PFGE, which has been used since 1984, is digestion of genomic DNA with rare cutter restriction enzymes specific for the organism of interest.18 For example, XbaI is the enzyme most commonly used for Salmonella serovars. After restriction enzymes digest intact genomic DNA embedded in agarose, digested fragments are separated in a pulsed-field gel electrophoresis apparatus. The most common system of electrophoresis is the contour-clamped homogenous electric field system (CHEF: BioRad Laboratories, Hercules, California). This method has been applied to identification of various pathogens and has been the standard genotyping technique adopted by the Center for Disease Control and Prevention (CDC) for subtyping seven foodborne pathogens. The CDC central database, also called Pulsenet,19 is an early warning system to detect unique PFGE patterns in isolates from cases of foodborne disease in humans and from food commodities. Detailed information on this system can be found at the Pulsenet website.19
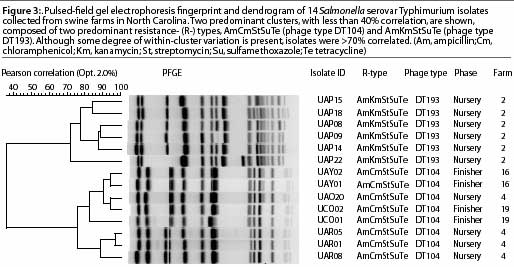
The standard PFGE protocol has been previously described.20 Figure 3 shows distinctly different cluster PFGE patterns within and between different R-types (ACSSuT and AKSSuT) and phage types (DT104 and DT193) of Salmonella isolates from swine farms.
PCR-based approaches: AFLP
Amplified fragment length polymorphism, a recently developed alternative fingerprinting method,21 is a PCR-based typing system technically different from PFGE, with a high-resolution approach and high throughput capacity. Like other PCR-based approaches, this method is sensitive to contamination that may affect its reproducibility, and subsequently its sensitivity and specificity. In addition, this method requires relatively more expensive equipment, ie, a DNA sequencer, which is not available in most research or field laboratories. In preliminary studies, the discriminatory power of this method was similar to that of PFGE (Gebreyes, unpublished data). Published studies have also reported similar findings.22,23 The evidence in these reports and the standard use of PFGE in the national database (Pulsenet) make PFGE the preferred choice for subtyping foodborne pathogens, particularly Salmonella. Figure 4 demonstrates the use of AFLP in subtyping 48 Salmonella isolates collected from swine in North Carolina. Four clusters with 60% or less pair-wise correlation coefficients are shown.

Other PCR-based methods, including Rep-PCR, have also been demonstrated as valuable tools for subtyping foodborne Salmonella from swine24 and other swine pathogens, such as Hemophilus parasuis.25 However, the discriminatory power and reproducibility of Rep-PCR is not well established and its use has been limited.
Conclusion
The described molecular diagnostic and subtyping methods have the potential to play a pivotal role in identification of foodborne pathogens at individual or population levels. Although methods based on singleplex PCR have been commonly used in diagnostic laboratories for diagnosis of clinical swine diseases, their use in identifying foodborne pathogens isolated from swine is limited, as these pathogens occur subclinically and their characterization is usually based on phenotyping (for example serotyping and phage typing of Salmonella). In research settings, PCR assays (traditional and real-time) are being further developed for rapid and accurate identification of foodborne pathogens in human foodborne disease outbreaks. Molecular methods in foodborne disease outbreak investigations are currently complemented with phenotypic analysis, because the genetic composition of foodborne strains remains largely unknown. One molecular method, PFGE, has gained wide acceptance for subtyping Salmonella isolated from humans because of the development of the Pulsenet database system. Previously, fingerprints of Salmonella isolated from swine and humans have been found to be identical and outbreak origin has been traced back to swine.26 In addition, human and swine Salmonella isolates from the same geographical location and with identical phenotypes (based on serotyping and antibiotyping) have also been distinguished (Gebreyes et al, unpublished data). As multiple genetic clones may exist within phenotypes, phenotyping is not sufficient to indicate clonality of organisms among various hosts. To fully understand the extent of strain sharing between different hosts, a fingerprint database for swine Salmonella (and other foodborne pathogens) must be developed and compared with the fingerprints of human isolates.
References – refereed
2. Oliveira SD, Santos LR, Schuch DM, Silva AB, Salle CT, Canal CW. Detection and identification of salmonellas from poultry-related samples by PCR. Vet Microbiol. 2002;87:25-35.
3. Gebreyes WA, Altier C. Molecular characterization of multidrug-resistant Salmonella enterica subsp. enterica serovar Typhimurium isolates from swine. J Clin Microbiol. 2002;40:2813-2822.
4. Heisig P, Kratz B, Halle E, Graser Y, Altwegg M, Rabsch W, Faber JP. Identification of DNA gyrase A mutations in ciprofloxacin-resistant isolates of Salmonella typhimurium from men and cattle in Germany. Microb Drug Resist. 1995;1:211-218.
5. Carlson SA, Bolton LF, Briggs CE, Hurd HS, Sharma VK, Fedorka-Cray PJ, Jones BD. Detection of multiresistant Salmonella typhimurium DT104
using multiplex and fluorogenic PCR. Mol Cell Probes. 1999;13:213-222.
6. Chen W, Martinez G, Mulchandani A. Molecular beacons: a real-time polymerase chain reaction assay for detecting Salmonella. Anal Biochem. 2000;280:166-172.
7. Doran JL, Collinson SK, Kay CM, Banser PA, Burian J, Munro CK, Lee SH, Somers JM, Todd EC, Kay WW. fimA and tetC based DNA diagnostics for Salmonella. Mol Cell Probes. 1994;8:291-310.
8. Liu D, Verma NK, Romana LK, Reeves PR. Relationships among the rfb regions of Salmonella serovars A, B, and D. J Bacteriol. 1991;173:4814-4819.
9. Brown PK, Romana LK, Reeves PR. Cloning of the rfb gene cluster of a group C2 Salmonella strain:
comparison with the rfb regions of groups B and D. Mol Microbiol. 1991;5:1873-1881.
11. Bakshi CS, Singh VP, Malik M, Sharma B, Singh RK. Polymerase chain reaction amplification of 16S-23S spacer region for rapid identification of Salmonella serovars. Acta Vet Hung. 2002;50:161-166.
12. Center for Disease Control and Prevention (CDC). Salmonella Annual Summary. 2000;1-15.
14. Briggs CE, Fratamico PM. Molecular characterization of an antibiotic resistance gene cluster of Salmonella typhimurium DT104. Antimicrob Agents Chemother. 1999;43:846-849.
15. Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986-994.
16. Chen S, Yee A, Griffiths M, Larkin C, Yamashiro CT, Behari R, Paszko-Kolva C, Rahn K, De Grandis SA. The evaluation of a fluorogenic polymerase chain reaction assay for the detection of Salmonella species in food commodities. Int J Food Microbiol. 1997;35:239-250.
18. Schwartz DC, Cantor CR. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell. 1984;37:67-75.
20. Gautom RK. Rapid pulsed-field gel electrophoresis protocol for typing of Escherichia coli O157:H7 and other gram-negative organisms in 1 day. J Clin Microbiol. 1997;35:2977-2980.
21. Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zadeau M. AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res. 1995;23:4407-4414.
22. Lindstedt BA, Heir E, Vardund T, Kapperud G. Fluorescent amplified-fragment length polymorphism genotyping of Salmonella enterica subsp. enterica serovars and comparison with pulsed-field gel electrophoresis typing. J Clin Microbiol. 2000;38:1623-1627.
23. Liebana E, Garcia-Migura L, Clouting C, Cassar CA, Clifton-Hadley FA, Lindsay EA, Threlfall EJ, Chappell SA, Davies RH. Investigation of the genetic diversity among isolates of Salmonella enterica serovar Dublin from animals and humans from England, Wales and Ireland. J Appl Microbiol. 2002; 93:732-744.
24. Weigel RM, Qiao B, Barber DA, Teferedegne B, Kocherginskaya S, White BA, Isaacson RE. Identification of patterns of transmission of Salmonella within swine production systems using pulsed field gel electrophoresis (PFGE) and repetitive sequence polymerase chain reaction (REP-PCR): a quantitative analysis. Berl Munch Tierarztl Wochenschr. 2001;114:397-400.
26. Hampton MD, Threlfall EJ, Frost JA, Ward LR, Rowe B. Salmonella typhimurium DT 193: differentiation of an epidemic phage type by antibiogram, plasmid profile, plasmid fingerprint and salmonella plasmid virulence (spv) gene probe. J Appl Bacteriol. 1995;78:402-408.
References – non refereed
1. PCR Animation. Dolan DNA Learning Center, Cold Springs Harbor Laboratory. http://www.dnalc.org/shockwave/pcranwhole.html. Accessed February 6, 2003.
10. McQuiston JR, Reed TS, Langford CA, Brenner F, Fields PI. Comparative sequencing and initial development of a DNA Enzyme Immunosorbent Assay (EIA) for serotyping of the Salmonella H-antigen alleles. Proc Intl Conf Emerg Inf Dis. Atlanta, Georgia. 2000;116.
13. United States Department of Agriculture. Animal and Plant Health Inspection Services. Shedding of Salmonella by Finisher Hogs in the U.S. Info Sheet. Veterinary Services. http://www.aphis.usda.gov/vs/ceah/cahm/Swine/sw95salm.htm. Accessed February 7, 2003.
17. Hybridization probes for the detection of nucleic acids in homogeneous solutions. http://molecular-beacons.org/protocol.html. Accessed February 7, 2003.
19. CDC. Foodborne and Diarrheal Diseases Branch. What is Pulsenet? http://www.cdc.gov/pulsenetwhat_is.htm. Accessed February 7, 2003.
25. Oliviera S, Pijon C. Diagnosis of Haemophilus parasuis in affected herds and use of epidemiological data to control diseases. J Swine Health Prod. 2002; 10:221-225.